|
Affinity Chromatography in Brief
Affinity chromatography separates proteins on the basis of a reversible interaction between a protein (or group of proteins)
and a specific ligand coupled to a chromatography matrix. The technique offers high selectivity, hence high resolution, and
usually high capacity for the protein of interest. Purification can be in the order of several thousand-fold and recoveries
of active material are generally very high.
Affinity chromatography is unique in purification technology since it is the only technique that enables the purification
of a biomolecule on the basis of its biological function or individual chemical structure. Purification that would otherwise
be time-consuming, difficult or even impossible using other techniques can often be easily achieved with affinity chromatography.
The technique can be used to separate active biomolecules from denatured or functionally different forms, to isolate pure
substances present at low concentration in large volumes of crude sample and also to remove specific contaminants. (Amersham
Biosciences, 2008)
Affinity chromatography is a liquid chromatographic technique that uses a "biologically related" agent as a
stationary phase for the purification or analysis of sample components. The retention of solutes in this method is based on
the specific, reversible interactions that are found in biological systems, such as the binding of an enzyme with a substrate
or an antibody with an antigen. These interactions are exploited in affinity chromatography by placing one of a pair of interacting
molecules onto or within a solid support and using this as a stationary phase. This immobilized molecule is known as the affinity
ligand and is what gives an affinity column the ability to bind to particular compounds in a sample.
Affinity chromatography is a valuable tool in areas such as biochemistry, pharmaceutical science, clinical chemistry,
and environmental testing, where it has been used for both the purification and analysis of compounds in complex sample mixtures.
The strong and relatively specific binding that characterizes many affinity ligands allows solutes to be quantitated or purified
by these ligands with little or no interferences from other sample components. Often, the solute of interest can be isolated
in one or two steps, with purification yields of one hundred to several thousand-fold being common. (Hage, 2005)
History of Affinity Chromatography
Chromatography is one of the most truly chemical methods for manipulating and understanding the natural world. It is an enabling
technology for a wide range of products and discoveries, including those of more than a half-dozen Nobel Prize winners. All
of the various forms of chromatography rely on differential solubility or adsorption of compounds to separate molecules between
a stationary phase and a mobile phase.
Affinity chromatography, developed in the 1930s, was first used to study enzymes and other proteins. This method relies
on the affinity of various biochemical compounds for one another, such as enzymes for their substrates or antibodies for their
antigens. Incorporating one of the compounds to a solid support allows it to serve in selective chromatographic adsorption
of the unique molecules for which it has affinity. It has become a staple of genetic engineering, vaccine production, and
basic metabolic research. Arne Wilhelm Tiselius, winner of the 1948 Nobel Prize, was a fundamental contributor to perfecting
affinity chromatography through his development of many gel types for specific biochemical adsorption.
Since the 1930s, Tiselius worked on developing electrophoresis as part of US research on human plasma. His techniques
proved fundamental to the development of electrochromatography, a catch-all term for any form of chromatography, such as capillary
electrophoresis, which uses electromotive force as an aid to separations. In the 1950s, Tiselius and co-workers adapted molecular
sieving chromatography - a means of sorting macromolecules by size and molecular weight - for the purification of a host of
biologically important compounds, both natural and synthetic.

|
| A chemist using column chromatographic apparatus in the mid-1950s |
These are just some of the many "minor" forms of chromatographic analysis, each with its own history. Despite the
relatively small number of people involved in chromatography's early days, it is difficult to name a facet of modern life
to which it has not contributed, whether as an analytical science, a preparative tool, or a regulatory technique - including
its role as a mainstay of the chemical industry. In 1990 surveys, gas-liquid chromatographs were the most frequently mentioned
analytical instruments for purchase. Chromatography - with its fundamental quest to separate and identify every kind of atom
and molecule, and with its diverse applications from agriculture to zoology, from petrochemicals to biopharmaceuticals, and
from simple salts to the most complex polymers - can indeed be considered the instrumental backbone of chemistry's claim to
be the central science. (Lesney, 1998)
Design of Affinity Chromatography
Affinity chromatography is one of the most powerful separation methods that the biochemical engineer has at his disposal.
Enzymes, receptors, antigens, antibodies, hormones, binding proteins, whole cells and other substances which possess a specific
reversible binding site are amenable to highly selective purification. In contrast to classical techniques such as gel filtration
or fractional precipitation, which rely on small differences in molecular size, isoelectric point, etc., affinity procedures
utilize specific chemical reactivity which very few if any of the contaminants will have in common with the desired product.
Hundreds of such separations have been devised and described in the literature within the past five years. However, since
the enzyme purification is normally only an intermediate step in an investigation, when one finds a method for a particular
enzyme, it is normally not an optimal scheme which one would employ on a large scale. In industrial scale preparations, minimal
loss, maximum speed, the smallest amount of (usually) expensive column packing, etc. are important practical considerations.
(Graves and Wu, 1979)
Some typical biological interactions, frequently used in affinity chromatography, are listed below:
- Enzyme - substrate analogue, inhibitor, cofactor.
- Antibody - antigen, virus, cell.
- Lectin - polysaccharide, glycoprotein, cell surface receptor, cell.
- Nucleic acid - complementary base sequence, histones, nucleic acid polymerase, nucleic acid binding protein.
- Hormone, vitamin - receptor, carrier protein.
- Glutathione - glutathione-S-transferase or GST fusion proteins.
- Metal ions - Poly (His) fusion proteins, native proteins with histidine, cysteine and/or tryptophan residues on their
surfaces.
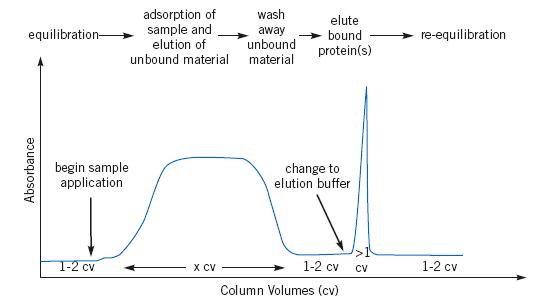
Stages in Affinity Chromatography
|

|
1. Affinity medium is equilibrated in binding buffer.
|
|

|
2. Sample is applied under conditions that favor specific binding of the target molecule(s) to a complementary binding substance
(the ligand). Target substances bind specifically, but reversibly, to the ligand and unbound material washes through the column.
|
|

|
3. Target protein is recovered by changing conditions to favor elution of the bound molecules. Elution performed specific,
using competitive ligand, or non-specific, by changing the pH, ionic strength or polarity. Target protein is collected in
a purified, concentrated form.
|
|

|
4. Affinity medium is re-equilibrated with binding buffer.
|
|
|
| (Biomedical Engineering, 2007) |
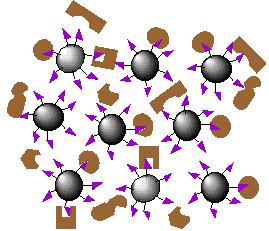
Affinity Chromatography Method
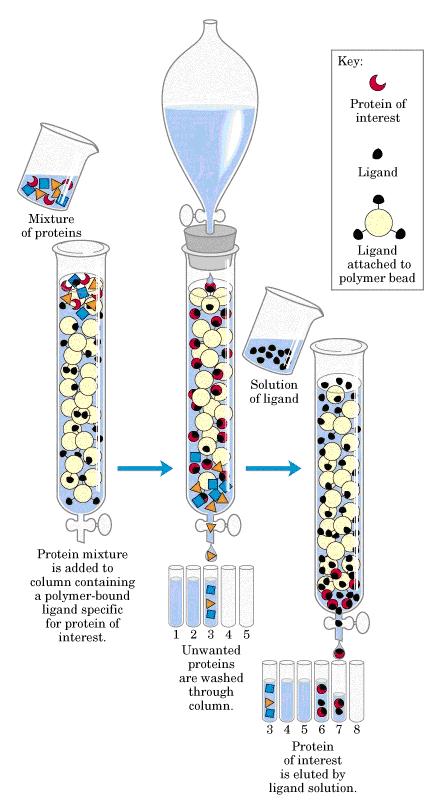
Affinity chromatography is designed to purify a particular protein from a mixed sample. (Davidson College, 2008)
|
| 1. Loading affinity column |
|
| 2. Proteins sieve through matrix of affinity beads |
|
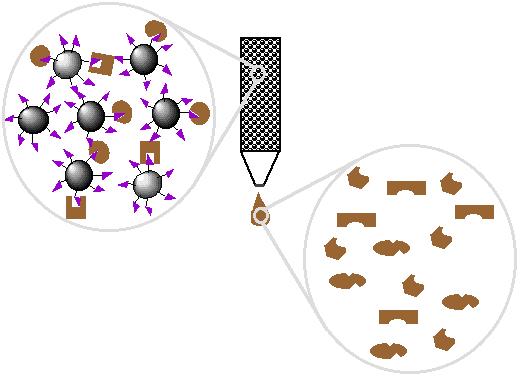
| 3. Proteins interact with affinity ligand with some binding loosely and others tightly |
|
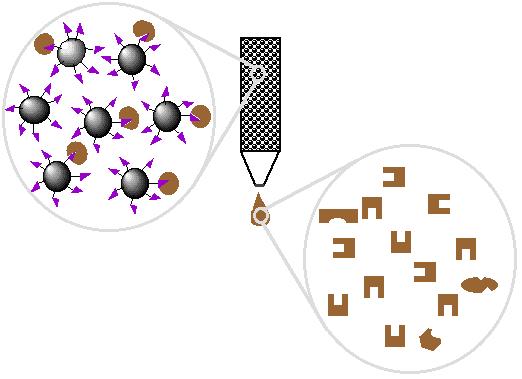
| 4. Wash off proteins that do not bind |
|
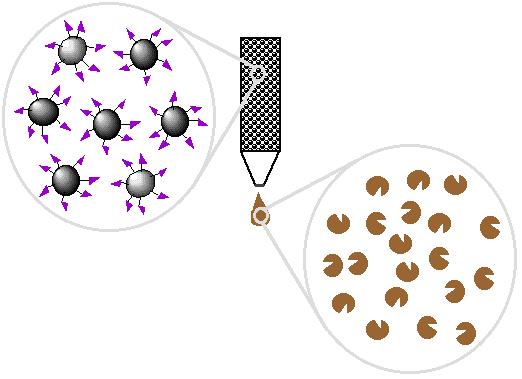
| 5. Wash off proteins that bind loosely |
|
| 6. Elute proteins that bind tightly to ligand and collect purified protein of interest |
Specific Uses of Affinity Chromatography
Affinity chromatography can be used in a number of applications, including nucleic acid purification, protein purification
from cell free extracts and antibody purification from blood serum. (Biomedical Engineering, 2007)
1.) Antibody affinity
Another use for the procedure is the affinity purification of antibodies from blood serum. If serum is known to contain
antibodies against a specific antigen (for example if the serum comes from an organism immunized against the antigen concerned)
then it can be used for the affinity purification of that antigen. For example if an organism is immunised against a GST-fusion
protein it will produce antibodies against the fusion-protein, and possibly antibodies against the GST tag as well. The protein
can then be covalently coupled to a solid support such as agarose.
For thoroughness the GST protein and the GST-fusion protein can each be coupled separately. The serum is initially allowed
to bind to the GST affinity matrix. This will remove antibodies against the GST part of the fusion protein. The serum is then
separated from the solid support and allowed to bind to the GST-fusion protein matrix. This allows any antibodies that recognize
the antigen to be captured on the solid support. Elution of the antibodies of interest is most often achieved using a low
pH buffer such as glycine pH 2.8. The eluate is collected into a neutral tris or phosphate buffer, to neutralize the low pH
elution buffer and halt any degradation of the antibody's activity. This is a nice example as affinity purification is used
to purify the initial GST-fusion protein, to remove the undesirable anti-GST antibodies from the serum and to purify the target
antibody.
2.) Immobilized metal ion affinity chromatography
Immobilized metal ion affinity chromatography (IMAC) is based on the specific coordinate covalent binding of amino acids,
allowing proteins with an affinity for metal ions to be retained in a column containing immobilized metal ions, such as cobalt,
nickel, copper, zinc, or iron ions. Many naturally occurring proteins do not have an affinity for metal ions, therefore recombinant
DNA techniques are used to introduce this property into a protein of interest. Methods used to elute the protein of interest
include changing the pH, or adding a specific molecule, such as imidazole. Similarly, the plant protein concanavalin A is
able to bind with glucose immobilised on a column matrix. The concavalin molecule is separated from the glucose by increasing
the glucose concentration, displacing the concavalin from the immobilised glucose.
3.) Recombinant proteins
Possibly the most common use of affinity chromatography is for the purification of recombinant proteins. Proteins with
a known affinity are tagged in order to aid their purification. The protein may have been genetically modified so as to allow
it to be selected for affinity binding; this is known as a fusion protein. Tags include His-tags and GST (glutathione-S-transferase)
tags. His6-tags have an affinity for nickel or cobalt ions which are coordinated with NTA for the purposes of solid medium
entrapment. For elution, an excess amount of a compound able to act as a nickel ligand, such as imidazole, is used. GST has
an affinity for glutathione - commercially available immobilized as glutathione agarose. For elution, excess glutathione is
used to displace the tagged protein.
Common Terms in Affinity Chromatography
Matrix: for ligand attachment. Matrix should be chemically and physically inert.
Spacer arm: used to improve binding between ligand and target molecule by overcoming any effects of steric hindrance.
Ligand: molecule that binds reversibly to a specific target molecule or group of target molecules.
Binding: buffer conditions are optimized to ensure that the target molecules interact effectively with the ligand and
are retained by the affinity medium as all other molecules wash through the column.
Elution: buffer conditions are changed to reverse (weaken) the interaction between the target molecules and the ligand
so that the target molecules can be eluted from the column.
Wash: buffer conditions that wash unbound substances from the column without eluting the target molecules or that re-equilibrate
the column back to the starting conditions (in most cases the binding buffer is used as a wash buffer).
Ligand coupling: covalent attachment of a ligand to a suitable pre-activated matrix to create an affinity medium.
Pre-activated matrices: matrices which have been chemically modified to facilitate the coupling of specific types of ligand.
Component of an Affinity Medium
The matrix
The matrix is an inert support to which a ligand can be directly or indirectly coupled. The list below highlights many
of the properties required for an efficient and effective chromatography matrix.
- Extremely low non-specific adsorption, essential since the success of affinity chromatography relies on specific interactions.
- Hydroxyl groups on the sugar residues are easily derivatized for covalent attachment of a ligand, providing an ideal
platform for the development of affinity media.
- An open pore structure ensures high capacity binding even for large biomolecules, since the interior of the matrix
is available for ligand attachment.
- Good flow properties for rapid separation.
- Stability under a range of experimental conditions such as high and low pH, detergents and dissociating agents.
Spacer arms
The binding site of a target protein is often located deep within the molecule and an affinity medium prepared by coupling
small ligands, such as enzyme cofactors, directly to Sepharose may exhibit low binding capacity due to steric interference.
Spacer arms must be designed to maximize binding, but to avoid non-specific binding effects.
The ligand
The ligand is the molecule that binds reversibly to a specific molecule or group of molecules, enabling purification by
affinity chromatography. The selection of the ligand for affinity chromatography is influenced by two factors:
- The ligand must exhibit specific and reversible binding affinity for the target substance(s)
- And it must have chemically modifiable groups that allow it to be attached to the matrix without destroying binding
activity.
|